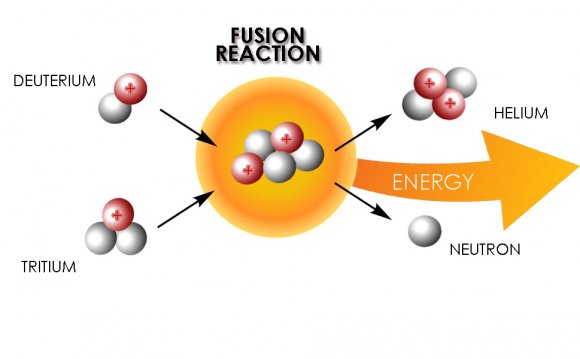
![Model of HIV-envelope-mediated membrane fusion according to Chan and Kim [4]. ...](/img/model_of_hiv_envelope_mediated_membrane_fusion_according_1.jpg) The current general model of HIV viral entry involves the binding of the trimeric viral envelope glycoprotein gp120/gp41 to cell surface receptor CD4 and chemokine co-receptor CXCR4 or CCR5, which triggers conformational changes in the envelope proteins. Gp120 then dissociates from gp41, allowing for the fusion peptide to be inserted into the target membrane and the pre-hairpin configuration of the ectodomain to form. The C-terminal heptad repeat region and the leucine/isoleucine zipper region then form the thermostable six-helix coiled-coil, which drives the membrane merger and eventual fusion. This model needs updating, as there has been a wealth of data produced in the last few years concerning HIV entry, including target cell dependencies, fusion kinetic data, and conformational intermediates. A more complete model must include the involvement of membrane microdomains, actin polymerization, glycosphingolipids, and possibly CD4 and chemokine signaling in entry. In addition, kinetic experiments involving the addition of fusion inhibitors have revealed some of the rate-limiting steps in this process, adding a temporal component to the model. A review of these data that may require an updated version of the original model is presented here.
The current general model of HIV viral entry involves the binding of the trimeric viral envelope glycoprotein gp120/gp41 to cell surface receptor CD4 and chemokine co-receptor CXCR4 or CCR5, which triggers conformational changes in the envelope proteins. Gp120 then dissociates from gp41, allowing for the fusion peptide to be inserted into the target membrane and the pre-hairpin configuration of the ectodomain to form. The C-terminal heptad repeat region and the leucine/isoleucine zipper region then form the thermostable six-helix coiled-coil, which drives the membrane merger and eventual fusion. This model needs updating, as there has been a wealth of data produced in the last few years concerning HIV entry, including target cell dependencies, fusion kinetic data, and conformational intermediates. A more complete model must include the involvement of membrane microdomains, actin polymerization, glycosphingolipids, and possibly CD4 and chemokine signaling in entry. In addition, kinetic experiments involving the addition of fusion inhibitors have revealed some of the rate-limiting steps in this process, adding a temporal component to the model. A review of these data that may require an updated version of the original model is presented here.
Keywords
- HIV;
- Fusion;
- Env;
- SIV;
- Virus;
- CD4;
- gp120;
- gp41;
- CXCR4;
- CCR5;
- Six-helix bundle
1. Introduction
HIV/SIV deliver their genetic material into the cell by direct fusion of the viral membrane with the plasma membrane of the host cells . The fusion is mediated by Env glycoproteins, , and, which are organized into oligomeric, probably trimeric spikes , and anchored in the viral membrane by the gp41 transmembrane protein. The Env glycoprotein, gp120, forms surface trimeric spikes, which are associated by noncovalent interactions with each subunit of the trimeric, normally hidden, gp41 . The similarity between structural motifs of gp120–gp41 and influenza hemagglutinin (HA) leads to the notion that the native conformation of gp41 is metastable and it is stabilized by gp120 and . The triggering mechanisms that activate Env are quite complex involving target cell CD4 , co-receptors, , , , and, and perhaps other cell surface components, and . These interactions trigger a barrage of conformational changes in Env that drive the membrane fusion process, , , , and (see ). The final outcome of the reaction is the formation of a six-helix bundled gp41 ectodomain core structure consisting of three N helices paired with three anti-parallel C helices, , and . The six-helix bundle structure is similar to the proposed fusogenic structures of envelope fusion proteins from influenza, Moloney murine leukemia virus, simian parainfluenza virus 5, Ebola virus, and simian immunodeficiency virus, as well as to the snarepin fusion machinery involved in intracellular fusion events . The formation of the six-helix bundle presumably occurs concomitantly with membrane fusion . HIV fusion can be inhibited by peptides that mimic the sequences of the N- and C-terminal helices by binding to the N terminal heptad repeat triple helices, or to the C-terminal regions of Env, thereby preventing six-helix bundle formation, and .
Fig. 1.
Model of HIV-envelope-mediated membrane fusion according to Chan and Kim . Upon binding to CD4 and co-receptor, gp120 undergoes conformational changes that allow gp41 fusion peptide insertion into the target membrane and the formation of a pre-hairpin structure. As the gp120 dissociates from gp41, the latter undergoes a slow reaction, which transforms the pre-hairpin into the six-helix bundle. It is during this process that the envelope-induced fusion is sensitive to inhibition by C-terminal peptides. (Current data show that Env–CD4 interactions trigger the pre-hairpin state that becomes sensitive to C-terminal inhibitors and that co-receptor engagement leads to rapid six-helix bundle formation ). The formation of the six-helix bundle promotes complete fusion, in which the fusion peptide and the transmembrane segment of gp41 lie parallel on a contiguous bilayer. Reprinted from , with permission from Elsevier.
YOU MIGHT ALSO LIKE



Share this Post
latest post
-
 Energy released in nuclear fusion October 31, 2017
Energy released in nuclear fusion October 31, 2017 -
 Nuclear fusion as an energy source October 29, 2017
Nuclear fusion as an energy source October 29, 2017 -
 Nuclear fusion clean energy October 27, 2017
Nuclear fusion clean energy October 27, 2017 -
 Order of magnitude estimate project Management October 25, 2017
Order of magnitude estimate project Management October 25, 2017 -
 Science Workshops October 23, 2017
Science Workshops October 23, 2017 -
 States of matter Plasma examples October 21, 2017
States of matter Plasma examples October 21, 2017 -
 What gas is the Sun made of? October 19, 2017
What gas is the Sun made of? October 19, 2017 -
 U.K. Scientists October 17, 2017
U.K. Scientists October 17, 2017 -
 Three orders of magnitude October 15, 2017
Three orders of magnitude October 15, 2017